Difference (from prior major revision)
Added: 0a1,2
> <b>What follows is a mostly complete description of the /practical/ steps required for performing a short molecular dynamics simulation of a DNA duplex. Please do keep in mind the difference between /doing/ things, and /understanding/ things.<b>
> -----------------
Changed: 6,27c8,21
< * *gunzip 1rop.pdb1.gz*
< * *mv 1rop.pdb1 dimer.pdb*
< * Edit the file /dimer.pdb/ and remove all lines that do not start with _ATOM_ or _TER_. Also, retain the final _END_. Now the difficult part : although the file contains two chains, these have the same identifier ' A '. You should find the beginning of the second chain and then use find-and-replace (or use a macro) to change _only for the second chain_ all occurences of the chain identifier ' A ' to ' B '. Save the modified file which should look like this :
< <code>
< ATOM 1 N MET A 1 31.007 2.290 18.034 1.00 28.97 N
< ATOM 2 CA MET A 1 32.390 2.582 17.546 1.00 26.82 C
< ATOM 3 C MET A 1 32.808 1.432 16.618 1.00 21.98 C
< ATOM 4 O MET A 1 32.375 0.280 16.846 1.00 23.60 O
< ......................
< ATOM 445 CE1 PHE A 56 38.342 8.335 16.790 1.00 25.49 C
< ATOM 446 CE2 PHE A 56 39.530 7.284 18.653 1.00 23.77 C
< ATOM 447 CZ PHE A 56 39.527 7.962 17.421 1.00 24.84 C
< TER 448 PHE A 56
< ATOM 1 N MET B 1 74.680 2.290 35.339 1.00 28.97 N
< ATOM 2 CA MET B 1 73.297 2.582 35.827 1.00 26.82 C
< ATOM 3 C MET B 1 72.879 1.432 36.755 1.00 21.98 C
< ATOM 4 O MET B 1 73.312 0.280 36.526 1.00 23.60 O
< ......................
< ATOM 446 CE2 PHE B 56 66.157 7.284 34.719 1.00 23.77 C
< ATOM 447 CZ PHE B 56 66.160 7.962 35.952 1.00 24.84 C
< TER 448 PHE B 56
< END
to
> * *gunzip 1BNA.pdb.gz*
> * *cp 1BNA.pdb ddoc.pdb*
> * Edit the file /ddoc.pdb/ and remove all lines that do not start with _ATOM_ or _TER_. Also, retain the final _END_. Save the modified file which should look like this :
> <code>
> ATOM 5 O5* C A 1 18.935 34.195 25.617 1.00 64.35 1BNA 67
> ATOM 6 C5* C A 1 19.130 33.921 24.219 1.00 44.69 1BNA 68
> ATOM 7 C4* C A 1 19.961 32.668 24.100 1.00 31.28 1BNA 69
> ATOM 8 O4* C A 1 19.360 31.583 24.852 1.00 37.45 1BNA 70
> ......
> ATOM 493 N2 G B 12 19.208 25.386 26.096 1.00 29.76 1BNA 551
> ATOM 494 N3 G B 12 18.350 23.438 27.053 1.00 21.95 1BNA 552
> ATOM 495 C4 G B 12 17.231 22.893 27.570 1.00 13.89 1BNA 553
> TER 496 G B 12 1BNA 554
> END 1BNA 700
Changed: 29c23
< * *rasmol dimer.pdb* (you should see the Rop dimer).
to
> * *rasmol ddoc.pdb* to look at it.
Changed: 31c25
to
Changed: 40c34
to
Added: 42a37
Changed: 45c40
to
Changed: 47c42
< * *rasmol aligned.pdb* (to confirm the changes)
to
> * *rasmol aligned.pdb* and then /set axes on/ (to confirm the changes).
Changed: 51c46
< ** Edit /A.pdb/ and retain only the A-chain coordinates. Add an _END_ at the end of the file. Find-and-replace 'HIS' with 'HSP' (fully protonated histidines, corresponding to performing a run at pH < 6{{For a run at neutral pH HIS should be replaced with either HSE or HSD depending on whether the histidine should be protonated on the δ or ε atoms. This may be determined from the hydrogen-bonding pattern of the histidine (in the context of the structure)}}). Save the file. Repeat for /B.pdb/. For example, /A.pdb/ should look like this :
to
> ** Edit /A.pdb/ and retain only the A-chain coordinates. Add an _END_ at the end of the file. Repeat for /B.pdb/. For example, /A.pdb/ should look like this :
Changed: 53,60c48,55
< ATOM 1 N MET A 1 22.545 -6.589 6.155 1.00 28.97 N
< ATOM 2 CA MET A 1 21.754 -5.355 5.863 1.00 26.82 C
< ATOM 3 C MET A 1 21.994 -4.366 7.013 1.00 21.98 C
< ......................
< ATOM 445 CE1 PHE A 56 17.522 -1.101 0.110 1.00 25.49 C
< ATOM 446 CE2 PHE A 56 15.440 -1.841 1.161 1.00 23.77 C
< ATOM 447 CZ PHE A 56 16.199 -0.871 0.483 1.00 24.84 C
< END
to
> ATOM 5 O5* C A 1 -18.727 -10.552 3.524 1.00 64.35 1BNA 67
> ATOM 6 C5* C A 1 -17.323 -10.651 3.227 1.00 44.69 1BNA 68
> ATOM 7 C4* C A 1 -17.096 -10.088 1.846 1.00 31.28 1BNA 69
> .........
> ATOM 245 N2 G A 12 18.548 0.358 -2.572 1.00 40.53 1BNA 307
> ATOM 246 N3 G A 12 18.570 -1.926 -2.048 1.00 37.34 1BNA 308
> ATOM 247 C4 G A 12 18.657 -2.793 -1.013 1.00 31.14 1BNA 309
> END
Changed: 62,63c57,58
< * Create a file with the name /psfgen.csh/ containing the following :
< <source>;
to
> * Create a file with the name /psfgen.csh/ containing the following (see the NAMD manual for psfgen documentaion. For more details on using psfgen with DNA see [http://www.ks.uiuc.edu/~villa/dna/]) :
> <code>
Added: 66a62,97
> alias residue C CYT
> alias residue T THY
> alias residue A ADE
> alias residue G GUA
> alias atom THY O5* O5'
> alias atom THY C5* C5'
> alias atom THY C4* C4'
> alias atom THY O4* O4'
> alias atom THY C1* C1'
> alias atom THY C2* C2'
> alias atom THY C3* C3'
> alias atom THY O3* O3'
> alias atom GUA O5* O5'
> alias atom GUA C5* C5'
> alias atom GUA C4* C4'
> alias atom GUA O4* O4'
> alias atom GUA C1* C1'
> alias atom GUA C2* C2'
> alias atom GUA C3* C3'
> alias atom GUA O3* O3'
> alias atom ADE O5* O5'
> alias atom ADE C5* C5'
> alias atom ADE C4* C4'
> alias atom ADE O4* O4'
> alias atom ADE C1* C1'
> alias atom ADE C2* C2'
> alias atom ADE C3* C3'
> alias atom ADE O3* O3'
> alias atom CYT O5* O5'
> alias atom CYT C5* C5'
> alias atom CYT C4* C4'
> alias atom CYT O4* O4'
> alias atom CYT C1* C1'
> alias atom CYT C2* C2'
> alias atom CYT C3* C3'
> alias atom CYT O3* O3'
Changed: 68c99,101
to
> first 5TER
> last 3TER
> pdb A.pdb
Changed: 70c103,114
to
> patch DEO2 A:2
> patch DEO2 A:4
> patch DEO2 A:5
> patch DEO2 A:6
> patch DEO2 A:10
> patch DEO2 A:12
> patch DEO1 A:1
> patch DEO1 A:3
> patch DEO1 A:7
> patch DEO1 A:8
> patch DEO1 A:9
> patch DEO1 A:11
Changed: 73c117,119
to
> first 5TER
> last 3TER
> pdb B.pdb
Changed: 75c121,132
to
> patch DEO2 B:2
> patch DEO2 B:4
> patch DEO2 B:5
> patch DEO2 B:6
> patch DEO2 B:10
> patch DEO2 B:12
> patch DEO1 B:1
> patch DEO1 B:3
> patch DEO1 B:7
> patch DEO1 B:8
> patch DEO1 B:9
> patch DEO1 B:11
Changed: 82,90c139,261
< </source>
< * Use it to generate a new pdb file (/psfgen.pdb/) and the PSF file (/psfgen.psf/) needed for NAMD : *source psfgen.csh*. Examine the output from the program (file /psfgen.log/).
< * Create a file with the name <i>make_all.vmd</i> containing the following script (for VMD) :
< <source>
< #
< # This is a short script implementing VMD's 'solvate' and
< # 'autoionize' to prepare a fully solvated and neutral
< # system for a periodic boundary simulation. It also
< # prepares two pdb files needed for implementing restrains
to
> </code>
> * Use it to generate a new pdb file (/psfgen.pdb/) and the PSF file (/psfgen.psf/) needed for the steps that follow : *source psfgen.csh*. Examine the output from the program (file /psfgen.log/).
> * We now need to neutralise the DNA charges. The simple-minded approach (with VMD's /autoionize/ plugin) used for proteins is not satisfactory for DNA : the ions' placement must take into account the DNA's electrostatic field. The best approach would have been to solve the Poisson-Boltzmann equation (with the program Delphi for example) and use the calculated field to guide ion placement. A more simple minded (and less computationally intensive) approach is to start placing the ions one-by-one using a simple Coulomb-based potential. This is implemented with the program /sodium/, do a <b>less /usr/local/doc/sodium.c</b> to read its documentation. The steps we need to take are :
> ** Prepare a PDB file containing partial charges on the B-factor column. This you do with a script this time for xplor. Save the following script in a file named /xplor.sh/, and run it with /source xplor.sh/.
> <code>
> #!/bin/tcsh -f
> xplorbig > xplor.log << eof
> structure @psfgen.psf end
> coor @psfgen.pdb
> vector do (B=CHARGE) (all)
> write coor output="psfgen_charges.pdb" end
> stop
> eof
> exit
> </code>
> ** Examine the output (file <i>psfgen_charges.pdb</i>) and confirm that the charges are there.
> ** Prepare a configuration file for /sodium/ with the name *sodium.config* containing the following :
> <code>
> NUM_IONS 22
> R_ION_PRODNA 6.5
> R_ION_ION 11.0
> GRID_STEP 0.50
> BORDER_WIDTH 10.0
> PDB_NAME psfgen_charges.pdb
> PDB_OUT ions.pdb
> </code>
> ** Run it <b>sodium < sodium.config > sodium.log</b>. For such a small system it will only take a minute or two.
> ** Check the /sodium.log/ file, check the ion distribution : <b>cat psfgen.pdb ions.pdb > sodium_test.pdb ; rasmol sodium_test.pdb</b> and then /select not DNA/, /cpk/.
> ** You can experiment by changing the /sodium/ parameters controling either the minimum distance between ions and DNA, or the minimum ion-ion distance. Compare the resulting structures to see how the ions structure changes. How would you choose values for these two parameters ?
> ** Re-create PDB and PSF files for the system this time including the ions. To do this prepare a file <i>psfgen_ions.sh</i> containing the following :
> <code>
> #!/bin/tcsh -f
> /usr/local/NAMD_2.5/psfgen >& psfgen_ions.log << END
> topology top_all27_prot_na.inp
> alias residue C CYT
> alias residue T THY
> alias residue A ADE
> alias residue G GUA
> alias atom THY O5* O5'
> alias atom THY C5* C5'
> alias atom THY C4* C4'
> alias atom THY O4* O4'
> alias atom THY C1* C1'
> alias atom THY C2* C2'
> alias atom THY C3* C3'
> alias atom THY O3* O3'
> alias atom GUA O5* O5'
> alias atom GUA C5* C5'
> alias atom GUA C4* C4'
> alias atom GUA O4* O4'
> alias atom GUA C1* C1'
> alias atom GUA C2* C2'
> alias atom GUA C3* C3'
> alias atom GUA O3* O3'
> alias atom ADE O5* O5'
> alias atom ADE C5* C5'
> alias atom ADE C4* C4'
> alias atom ADE O4* O4'
> alias atom ADE C1* C1'
> alias atom ADE C2* C2'
> alias atom ADE C3* C3'
> alias atom ADE O3* O3'
> alias atom CYT O5* O5'
> alias atom CYT C5* C5'
> alias atom CYT C4* C4'
> alias atom CYT O4* O4'
> alias atom CYT C1* C1'
> alias atom CYT C2* C2'
> alias atom CYT C3* C3'
> alias atom CYT O3* O3'
> segment A {
> first 5TER
> last 3TER
> pdb A.pdb
> }
> patch DEO2 A:2
> patch DEO2 A:4
> patch DEO2 A:5
> patch DEO2 A:6
> patch DEO2 A:10
> patch DEO2 A:12
> patch DEO1 A:1
> patch DEO1 A:3
> patch DEO1 A:7
> patch DEO1 A:8
> patch DEO1 A:9
> patch DEO1 A:11
> coordpdb A.pdb A
> segment B {
> first 5TER
> last 3TER
> pdb B.pdb
> }
> patch DEO2 B:2
> patch DEO2 B:4
> patch DEO2 B:5
> patch DEO2 B:6
> patch DEO2 B:10
> patch DEO2 B:12
> patch DEO1 B:1
> patch DEO1 B:3
> patch DEO1 B:7
> patch DEO1 B:8
> patch DEO1 B:9
> patch DEO1 B:11
> coordpdb B.pdb B
> segment SOD {
> pdb ions.pdb
> }
> coordpdb ions.pdb SOD
> guesscoord
> writepsf psfgen_ions.psf
> writepdb psfgen_ions.pdb
> END
> exit
> </code>
> ** Run it with <b>source psfgen_ions.sh</b>, examine log and PDB file.
> * Time to hydrate the system : Create a file with the name <i>vmd.script</i> containing the following script (for VMD) :
> <code>
> #
> # This is a short script implementing VMD's 'solvate' to prepare
> # a fully solvated system for a periodic boundary simulation.
> # It also prepares two pdb files needed for implementing restrains
Changed: 96c267
< solvate psfgen.psf psfgen.pdb -o hydrated -b 1.80 -t 15.0
to
> solvate psfgen_ions.psf psfgen_ions.pdb -o hydrated -b 1.80 -t 15.0
Changed: 98c269,271
< # Add ions to neutralise charge
to
> # Here you can add additional ions to emulate higher ionic strength
> # conditions. In this case we add 50mM salt, increasing the effective
> # ionic strength to approximately 250mM.
Changed: 107c280
< set sel [atomselect top "protein and name CA"]
to
> set sel [atomselect top "nucleic and name P"]
Changed: 110c283
< $all writepdb restrain_ca.pdb
to
> $all writepdb restrain_P.pdb
Changed: 112c285
< set to_fix [atomselect top "protein and backbone"]
to
> set to_fix [atomselect top "nucleic and backbone"]
Changed: 116c289
to
Changed: 120,121c293,294
< ** In the console give <b>source make_all.vmd</b>
< ** If all goes well you should see your fully hydrated and ionised system on VMD's graphics window.
to
> ** In the console give <b>source vmd.script</b>
> ** If all goes well you should see your fully hydrated and ionised system on VMD's graphics window{{VMD's selection keyword 'nucleic' requires the presence of phosphorus atoms. The result is that when selecting with the 'nucleic' keyword the 5' termini will *not* show-up. You can avoid this by saying something like 'not (water or ions)'}}.
Added: 122a296
> * Use /rasmol/ or /vmd/ to study your system (ionized.pdb). Make sure that there are no waters or ions accidentally placed inside your molecule's hydrophobic core.
Changed: 130,132c304,306
< On X : -39.30 39.30 0.00 78.60
< On Y : -30.05 30.05 0.00 60.10
< On Z : -25.98 30.85 2.44 56.83
to
> On X : -37.70 36.29 -0.71 74.00
> On Y : -29.96 30.01 0.03 59.97
> On Z : -30.39 29.60 -0.39 60.00
Changed: 134c308
< Note these numbers, especially the range (on x,y,z) and position of centre.
to
> ** Note these numbers, especially the range (on x,y,z) and position of centre.
Changed: 136c310
to
Changed: 166,167c340,341
< nonBondedFreq 2
< fullElectFrequency 4
to
> nonBondedFreq 1
> fullElectFrequency 2
Changed: 189c363
< PmeGridsizeY 64 # <===== CHANGE ME
to
> PmeGridsizeY 60 # <===== CHANGE ME
Changed: 196,199c370,373
< cellBasisVector1 78.60 00.00 00.00 # <===== CHANGE ME
< cellBasisVector2 00.00 60.10 00.00 # <===== CHANGE ME
< cellBasisVector3 00.00 00.00 56.83 # <===== CHANGE ME
< cellOrigin 0.00 0.00 2.44 # <===== CHANGE ME
to
> cellBasisVector1 74.00 00.00 00.00 # <===== CHANGE ME
> cellBasisVector2 00.00 60.00 00.00 # <===== CHANGE ME
> cellBasisVector3 00.00 00.00 60.00 # <===== CHANGE ME
> cellOrigin 0.00 0.00 0.00 # <===== CHANGE ME
Changed: 211,212c385,386
< consRef restrain_ca.pdb
< consKFile restrain_ca.pdb
to
> consRef restrain_P.pdb
> consKFile restrain_P.pdb
Changed: 252c426
< # heat with CAs restrained
to
> # heat with Ps restrained
Changed: 258c432
to
Changed: 262c436
< # equilibrate volume with CAs restrained
to
> # equilibrate volume with Ps restrained
Changed: 266c440
to
Changed: 272c446
to
Changed: 274c448,450
< ** Type *mosmon* from your terminal. You should see something similar to this [[image: Mosmon, three idle nodes]] This graph shows (in real time) the CPU load for each of the nodes that are currently part of the cluster. The horizontal axis shows the various nodes, the vertical axis shows the corresponding load. If all machines are working properly /and they are all running the openMosix linux kernel/ you should see at least 18 nodes (numbered 25700-25716, plus another node with ID 25854).
to
> ** Type *mosmon* from your terminal. You should see something similar to this
> [[image: Mosmon, three idle nodes]]
> ** This graph shows (in real time) the CPU load for each of the nodes that are currently part of the cluster. The horizontal axis shows the various nodes, the vertical axis shows the corresponding load. If all machines are working properly /and they are all running the openMosix linux kernel/ you should see at least 18 nodes (numbered 25700-25716, plus another node with ID 25854).
Changed: 276c452,456
< ** Type *q* to stop mosmon and then give *qmon* (from your terminal). The main Sun Grid Engine GUI window should appear: [[image: SGE qmon main window]] Select "Queue control". This should pop-up the corresponding window : [[image: SGE qmon qcontrol window]] Note the small green boxes (indicating that a job is running) on all nodes except pc08, pc09 and pc10. So, it does look like we could use these three machines for the tests. Press 'Done' on the queue control window and then exit in the qmon window.
to
> ** Type *q* to stop mosmon and then give *qmon* (from your terminal). The main Sun Grid Engine GUI window should appear:
> [[image: SGE qmon main window]]
> ** Select "Queue control". This should pop-up the corresponding window :
> [[image: SGE qmon qcontrol window]]
> ** Note the small green boxes (indicating that a job is running) on all nodes except pc08, pc09 and pc10. So, it does look like we could use these three machines for the tests. Press 'Done' on the queue control window and then exit in the qmon window.
Changed: 285c465
< cp ionized.p* par_all27_prot_na.inp heat.namd fix_backbone.pdb restrain_ca.pdb /work/tmp/my_tests/
to
> cp ionized.p* par_all27_prot_na.inp heat.namd fix_backbone.pdb restrain_P.pdb /work/tmp/my_tests/
Changed: 305c485
< Info: 1 NAMD 2.5 Linux-i686 1 pc09.cluster.mbg.gr glykos
to
> Info: 1 NAMD 2.5 Linux-i686 1 aspera.cluster.mbg.gr glykos
Changed: 315,318c495,498
< Info: PERIODIC CELL BASIS 1 79.6 0 0
< Info: PERIODIC CELL BASIS 2 0 61.1 0
< Info: PERIODIC CELL BASIS 3 0 0 57.83
< Info: PERIODIC CELL CENTER 0 0 2.44
to
> Info: PERIODIC CELL BASIS 1 74 0 0
> Info: PERIODIC CELL BASIS 2 0 60 0
> Info: PERIODIC CELL BASIS 3 0 0 60
> Info: PERIODIC CELL CENTER 0 0 0
Changed: 381c561
< Info: PME GRID DIMENSIONS 80 64 60
to
> Info: PME GRID DIMENSIONS 80 60 60
Changed: 394c574
< Info: RANDOM NUMBER SEED 1099129124
to
> Info: RANDOM NUMBER SEED 1109584307
Changed: 409,413c589,593
< Info: 25368 ATOMS
< Info: 17518 BONDS
< Info: 11126 ANGLES
< Info: 4764 DIHEDRALS
< Info: 308 IMPROPERS
to
> Info: 25117 ATOMS
> Info: 17038 BONDS
> Info: 9599 ANGLES
> Info: 2228 DIHEDRALS
> Info: 68 IMPROPERS
Changed: 415,417c595,597
< Info: 112 CONSTRAINTS
< Info: 450 FIXED ATOMS
< Info: 24450 RIGID BONDS
to
> Info: 22 CONSTRAINTS
> Info: 176 FIXED ATOMS
> Info: 24569 RIGID BONDS
Changed: 419,423c599,603
< Info: 50304 DEGREES OF FREEDOM
< Info: 8768 HYDROGEN GROUPS
< Info: 226 HYDROGEN GROUPS WITH ALL ATOMS FIXED
< Info: TOTAL MASS = 154680 amu
< Info: TOTAL CHARGE = 1.60187e-06 e
to
> Info: 50254 DEGREES OF FREEDOM
> Info: 8647 HYDROGEN GROUPS
> Info: 108 HYDROGEN GROUPS WITH ALL ATOMS FIXED
> Info: TOTAL MASS = 154535 amu
> Info: TOTAL CHARGE = 8.23289e-07 e
Changed: 425,428c605,608
< Info: Entering startup phase 0 with 7505 kB of memory in use.
< Info: Entering startup phase 1 with 7505 kB of memory in use.
< Info: Entering startup phase 2 with 9341 kB of memory in use.
< Info: Entering startup phase 3 with 9541 kB of memory in use.
to
> Info: Entering startup phase 0 with 7301 kB of memory in use.
> Info: Entering startup phase 1 with 7301 kB of memory in use.
> Info: Entering startup phase 2 with 8984 kB of memory in use.
> Info: Entering startup phase 3 with 9184 kB of memory in use.
Changed: 431,432c611,612
< Info: LARGEST PATCH (17) HAS 822 ATOMS
< Info: Entering startup phase 4 with 12754 kB of memory in use.
to
> Info: LARGEST PATCH (5) HAS 760 ATOMS
> Info: Entering startup phase 4 with 12394 kB of memory in use.
Changed: 439,441c619,621
< Info: Entering startup phase 5 with 14006 kB of memory in use.
< Info: Entering startup phase 6 with 14006 kB of memory in use.
< Info: Entering startup phase 7 with 14006 kB of memory in use.
to
> Info: Entering startup phase 5 with 13567 kB of memory in use.
> Info: Entering startup phase 6 with 13567 kB of memory in use.
> Info: Entering startup phase 7 with 13567 kB of memory in use.
Changed: 447,448c627,628
< Info: Entering startup phase 8 with 17392 kB of memory in use.
< Info: Finished startup with 22280 kB of memory in use.
to
> Info: Entering startup phase 8 with 16909 kB of memory in use.
> Info: Finished startup with 21797 kB of memory in use.
Changed: 451c631
< ENERGY: 0 7954.7376 5748.6073 437.1674 41.0044 -82921.4480 14265.3625 0.0000 0.0000 0.0000 -54474.5687 0.0000 -54474.5687 -54474.5687 0.0000 5922.6194 7853.0443 281259.6748 5922.6194 7853.0443
to
> ENERGY: 0 8367.1615 2562.3243 745.3077 0.7495 -86828.1909 2661304.0749 0.0000 0.0000 0.0000 2586151.4269 0.0000 2586151.4269 2586151.4269 0.0000 2770784.8777 2821435.1130 266400.0000 2770784.8777 2821435.1130
Changed: 455c635
< ENERGY: 0 7954.7376 5748.6073 437.1674 41.0044 -82921.4480 14265.3625 0.0000 0.0000 0.0000 -54474.5687 0.0000 -54474.5687 -54474.5687 0.0000 5922.6194 7853.0443 281259.6748 5922.6194 7853.0443
to
> ENERGY: 0 8367.1615 2562.3243 745.3077 0.7495 -86828.1909 2661304.0749 0.0000 0.0000 0.0000 2586151.4269 0.0000 2586151.4269 2586151.4269 0.0000 2770784.8777 2821435.1130 266400.0000 2770784.8777 2821435.1130
Changed: 457,463c637,642
< GRADIENT TOLERANCE: 28662.8
< ENERGY: 1 7935.4448 5745.3453 437.0249 40.9796 -82926.1450 14017.9693 0.0000 0.0000 0.0000 -54749.3812 0.0000 -54749.3812 -54749.3812 0.0000 5666.8255 7600.7423 281259.6748 5666.8255 7600.7423
< ENERGY: 2 7916.2985 5742.1035 436.8822 40.9549 -82930.8147 13791.9735 0.0000 0.0000 0.0000 -55002.6022 0.0000 -55002.6022 -55002.6022 0.0000 5432.3391 7370.3167 281259.6748 5432.3391 7370.3167
< ENERGY: 3 7878.4431 5735.6817 436.5970 40.9058 -82940.0682 13393.6985 0.0000 0.0000 0.0000 -55454.7421 0.0000 -55454.7421 -55454.7421 0.0000 5016.8834 6964.2358 281259.6748 5016.8834 6964.2358
< ENERGY: 4 7804.4690 5723.0900 436.0305 40.8087 -82958.2742 12757.4610 0.0000 0.0000 0.0000 -56196.4151 0.0000 -56196.4151 -56196.4151 0.0000 4345.9939 6314.8795 281259.6748 4345.9939 6314.8795
< ENERGY: 5 7663.3786 5698.9262 434.9535 40.6193 -82993.6089 11864.4871 0.0000 0.0000 0.0000 -57291.2443 0.0000 -57291.2443 -57291.2443 0.0000 3384.6148 5398.6693 281259.6748 3384.6148 5398.6693
< ENERGY: 6 7408.1034 5654.2007 433.5232 40.2601 -83060.8871 10739.2295 0.0000 0.0000 0.0000 -58785.5701 0.0000 -58785.5701 -58785.5701 0.0000 2131.4370 4224.4109 281259.6748 2131.4370 4224.4109
to
> GRADIENT TOLERANCE: 6.07456e+07
> ENERGY: 1 8461.3964 2575.7790 745.3495 0.7474 -86744.8887 66834.9357 0.0000 0.0000 0.0000 -8126.6806 0.0000 -8126.6806 -8126.6806 0.0000 62244.7987 65640.3834 266400.0000 62244.7987 65640.3834
> ENERGY: 2 8797.5006 2631.6879 745.3945 0.7454 -86710.0767 39177.7886 0.0000 0.0000 0.0000 -35356.9597 0.0000 -35356.9597 -35356.9597 0.0000 33177.5662 35521.3466 266400.0000 33177.5662 35521.3466
> ENERGY: 3 10026.8780 2749.8692 745.4941 0.7418 -86681.3159 31642.7422 0.0000 0.0000 0.0000 -41515.5906 0.0000 -41515.5906 -41515.5906 0.0000 24842.4717 28926.6604 266400.0000 24842.4717 28926.6604
> ENERGY: 4 16301.5980 3252.9024 745.7321 0.7358 -86621.8424 6474574.1627 0.0000 0.0000 0.0000 6408253.2887 0.0000 6408253.2887 6408253.2887 0.0000 6747235.8916 6748522.4575 266400.0000 6747235.8916 6748522.4575
> BRACKET: 6e-06 6.44977e+06 -8.35686e+09 2.51346e+09 1.63163e+09
Changed: 466c645
to
Added: 490a670,673
> # Nodes that can server as master queues
> #
> #$ -masterq pc01.q,pc02.q,pc03.q,pc04.q,pc05.q,pc06.q,pc07.q,pc08.q,pc09.q,pc10.q,pc11.q,pc12.q,pc13.q,pc14.q,pc16.q
> #
Changed: 513c696
to
Changed: 519c702,704
< * Type *mknamdplot LOG* and then click *Draw* to see a collection of graphs showing the evolution of quantities like the system's total energy, pressure, temperature, bond and angle energies, ... : [[image: mknamdplot example]] Click *Exit* to exit.
to
> * Type *mknamdplot LOG* and then click *Draw* to see a collection of graphs showing the evolution of quantities like the system's total energy, pressure, temperature, bond and angle energies, ... :
> [[image: mknamdplot example]]
> ** Click *Exit* to exit.
Changed: 523,531c708,716
< -rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:19 equil_ca.coor
< -rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:19 equil_ca.vel
< -rw-rw-r-- 1 glykos glykos 222 Oct 30 15:19 equil_ca.xsc
< -rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:16 heat_ca.coor
< -rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:13 heat_ca.coor.BAK
< -rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:16 heat_ca.vel
< -rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:13 heat_ca.vel.BAK
< -rw-rw-r-- 1 glykos glykos 153 Oct 30 15:16 heat_ca.xsc
< -rw-rw-r-- 1 glykos glykos 153 Oct 30 15:13 heat_ca.xsc.BAK
to
> -rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:19 equil_P.coor
> -rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:19 equil_P.vel
> -rw-rw-r-- 1 glykos glykos 222 Oct 30 15:19 equil_P.xsc
> -rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:16 heat_P.coor
> -rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:13 heat_P.coor.BAK
> -rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:16 heat_P.vel
> -rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:13 heat_P.vel.BAK
> -rw-rw-r-- 1 glykos glykos 153 Oct 30 15:16 heat_P.xsc
> -rw-rw-r-- 1 glykos glykos 153 Oct 30 15:13 heat_P.xsc.BAK
Changed: 563c748
to
Changed: 595,596c780,781
< nonBondedFreq 2
< fullElectFrequency 4
to
> nonBondedFreq 1
> fullElectFrequency 2
Changed: 617c802
< PmeGridsizeY 64 # <===== CHANGE ME
to
> PmeGridsizeY 60 # <===== CHANGE ME
Changed: 635c820
< langevinPistonDecay 500
to
> langevinPistonDecay 100
Changed: 640c825
to
Changed: 642c827
to
Added: 661a847,850
> # Nodes that can server as master queues
> #
> #$ -masterq pc01.q,pc02.q,pc03.q,pc04.q,pc05.q,pc06.q,pc07.q,pc08.q,pc09.q,pc10.q,pc11.q,pc12.q,pc13.q,pc14.q,pc16.q
> #
Changed: 681c870
to
What follows is a mostly complete description of the practical steps required for performing a short molecular dynamics simulation of a DNA duplex. Please do keep in mind the difference between doing things, and understanding things.
- Create a clean directory and cd in it.
- Get the CHARMM force field parameter and topology files :
- cp /usr/local/charmm27/par_all27_prot_na.inp ./
- cp /usr/local/charmm27/top_all27_prot_na.inp ./
- Download the coordinates for Dickerson's dodecamer from PDB [1] and save it on your disk.
- Edit the file ddoc.pdb and remove all lines that do not start with ATOM or TER. Also, retain the final END. Save the modified file which should look like this :
ATOM 5 O5* C A 1 18.935 34.195 25.617 1.00 64.35 1BNA 67
ATOM 6 C5* C A 1 19.130 33.921 24.219 1.00 44.69 1BNA 68
ATOM 7 C4* C A 1 19.961 32.668 24.100 1.00 31.28 1BNA 69
ATOM 8 O4* C A 1 19.360 31.583 24.852 1.00 37.45 1BNA 70
......
ATOM 493 N2 G B 12 19.208 25.386 26.096 1.00 29.76 1BNA 551
ATOM 494 N3 G B 12 18.350 23.438 27.053 1.00 21.95 1BNA 552
ATOM 495 C4 G B 12 17.231 22.893 27.570 1.00 13.89 1BNA 553
TER 496 G B 12 1BNA 554
END 1BNA 700
- rasmol ddoc.pdb to look at it.
- Now it is time to align the axes of inertia of the molecule with the orthogonal frame : prepare a file with the name moleman.csh containing the following :
#!/bin/tcsh -f
#
# This will read a PDB file and rotate/translate it so that
# 1. the centre of gravity will be at the origin
# 2. the axes of inertia will be aligned with the orthogonal frame
#
lx_moleman2 >& moleman.log << eof
/usr/local/xutil/moleman2.lib
REad ddoc.pdb
XYz ALign_inertia_axes
WRite aligned.pdb
quit
eof
exit
- Run it : source moleman.csh. This should create two files : a log file (moleman.log) and the new pdb (aligned.pdb).
- rasmol aligned.pdb and then set axes on (to confirm the changes).
- We need to prepare two pdb files each containing the coordinates of each of the two chains. The simplest approach is :
- Edit A.pdb and retain only the A-chain coordinates. Add an END at the end of the file. Repeat for B.pdb. For example, A.pdb should look like this :
ATOM 5 O5* C A 1 -18.727 -10.552 3.524 1.00 64.35 1BNA 67
ATOM 6 C5* C A 1 -17.323 -10.651 3.227 1.00 44.69 1BNA 68
ATOM 7 C4* C A 1 -17.096 -10.088 1.846 1.00 31.28 1BNA 69
.........
ATOM 245 N2 G A 12 18.548 0.358 -2.572 1.00 40.53 1BNA 307
ATOM 246 N3 G A 12 18.570 -1.926 -2.048 1.00 37.34 1BNA 308
ATOM 247 C4 G A 12 18.657 -2.793 -1.013 1.00 31.14 1BNA 309
END
- Create a file with the name psfgen.csh containing the following (see the NAMD manual for psfgen documentaion. For more details on using psfgen with DNA see [2]) :
#!/bin/tcsh -f
/usr/local/NAMD_2.5/psfgen >& psfgen.log << END
topology top_all27_prot_na.inp
alias residue C CYT
alias residue T THY
alias residue A ADE
alias residue G GUA
alias atom THY O5* O5'
alias atom THY C5* C5'
alias atom THY C4* C4'
alias atom THY O4* O4'
alias atom THY C1* C1'
alias atom THY C2* C2'
alias atom THY C3* C3'
alias atom THY O3* O3'
alias atom GUA O5* O5'
alias atom GUA C5* C5'
alias atom GUA C4* C4'
alias atom GUA O4* O4'
alias atom GUA C1* C1'
alias atom GUA C2* C2'
alias atom GUA C3* C3'
alias atom GUA O3* O3'
alias atom ADE O5* O5'
alias atom ADE C5* C5'
alias atom ADE C4* C4'
alias atom ADE O4* O4'
alias atom ADE C1* C1'
alias atom ADE C2* C2'
alias atom ADE C3* C3'
alias atom ADE O3* O3'
alias atom CYT O5* O5'
alias atom CYT C5* C5'
alias atom CYT C4* C4'
alias atom CYT O4* O4'
alias atom CYT C1* C1'
alias atom CYT C2* C2'
alias atom CYT C3* C3'
alias atom CYT O3* O3'
segment A {
first 5TER
last 3TER
pdb A.pdb
}
patch DEO2 A:2
patch DEO2 A:4
patch DEO2 A:5
patch DEO2 A:6
patch DEO2 A:10
patch DEO2 A:12
patch DEO1 A:1
patch DEO1 A:3
patch DEO1 A:7
patch DEO1 A:8
patch DEO1 A:9
patch DEO1 A:11
coordpdb A.pdb A
segment B {
first 5TER
last 3TER
pdb B.pdb
}
patch DEO2 B:2
patch DEO2 B:4
patch DEO2 B:5
patch DEO2 B:6
patch DEO2 B:10
patch DEO2 B:12
patch DEO1 B:1
patch DEO1 B:3
patch DEO1 B:7
patch DEO1 B:8
patch DEO1 B:9
patch DEO1 B:11
coordpdb B.pdb B
guesscoord
writepsf psfgen.psf
writepdb psfgen.pdb
END
exit
- Use it to generate a new pdb file (psfgen.pdb) and the PSF file (psfgen.psf) needed for the steps that follow : source psfgen.csh. Examine the output from the program (file psfgen.log).
- We now need to neutralise the DNA charges. The simple-minded approach (with VMD's autoionize plugin) used for proteins is not satisfactory for DNA : the ions' placement must take into account the DNA's electrostatic field. The best approach would have been to solve the Poisson-Boltzmann equation (with the program Delphi for example) and use the calculated field to guide ion placement. A more simple minded (and less computationally intensive) approach is to start placing the ions one-by-one using a simple Coulomb-based potential. This is implemented with the program sodium, do a less /usr/local/doc/sodium.c to read its documentation. The steps we need to take are :
- Prepare a PDB file containing partial charges on the B-factor column. This you do with a script this time for xplor. Save the following script in a file named xplor.sh, and run it with source xplor.sh.
#!/bin/tcsh -f
xplorbig > xplor.log << eof
structure @psfgen.psf end
coor @psfgen.pdb
vector do (B=CHARGE) (all)
write coor output="psfgen_charges.pdb" end
stop
eof
exit
- Examine the output (file psfgen_charges.pdb) and confirm that the charges are there.
- Prepare a configuration file for sodium with the name sodium.config containing the following :
NUM_IONS 22
R_ION_PRODNA 6.5
R_ION_ION 11.0
GRID_STEP 0.50
BORDER_WIDTH 10.0
PDB_NAME psfgen_charges.pdb
PDB_OUT ions.pdb
- Run it sodium < sodium.config > sodium.log. For such a small system it will only take a minute or two.
- Check the sodium.log file, check the ion distribution : cat psfgen.pdb ions.pdb > sodium_test.pdb ; rasmol sodium_test.pdb and then select not DNA, cpk.
- You can experiment by changing the sodium parameters controling either the minimum distance between ions and DNA, or the minimum ion-ion distance. Compare the resulting structures to see how the ions structure changes. How would you choose values for these two parameters ?
- Re-create PDB and PSF files for the system this time including the ions. To do this prepare a file psfgen_ions.sh containing the following :
#!/bin/tcsh -f
/usr/local/NAMD_2.5/psfgen >& psfgen_ions.log << END
topology top_all27_prot_na.inp
alias residue C CYT
alias residue T THY
alias residue A ADE
alias residue G GUA
alias atom THY O5* O5'
alias atom THY C5* C5'
alias atom THY C4* C4'
alias atom THY O4* O4'
alias atom THY C1* C1'
alias atom THY C2* C2'
alias atom THY C3* C3'
alias atom THY O3* O3'
alias atom GUA O5* O5'
alias atom GUA C5* C5'
alias atom GUA C4* C4'
alias atom GUA O4* O4'
alias atom GUA C1* C1'
alias atom GUA C2* C2'
alias atom GUA C3* C3'
alias atom GUA O3* O3'
alias atom ADE O5* O5'
alias atom ADE C5* C5'
alias atom ADE C4* C4'
alias atom ADE O4* O4'
alias atom ADE C1* C1'
alias atom ADE C2* C2'
alias atom ADE C3* C3'
alias atom ADE O3* O3'
alias atom CYT O5* O5'
alias atom CYT C5* C5'
alias atom CYT C4* C4'
alias atom CYT O4* O4'
alias atom CYT C1* C1'
alias atom CYT C2* C2'
alias atom CYT C3* C3'
alias atom CYT O3* O3'
segment A {
first 5TER
last 3TER
pdb A.pdb
}
patch DEO2 A:2
patch DEO2 A:4
patch DEO2 A:5
patch DEO2 A:6
patch DEO2 A:10
patch DEO2 A:12
patch DEO1 A:1
patch DEO1 A:3
patch DEO1 A:7
patch DEO1 A:8
patch DEO1 A:9
patch DEO1 A:11
coordpdb A.pdb A
segment B {
first 5TER
last 3TER
pdb B.pdb
}
patch DEO2 B:2
patch DEO2 B:4
patch DEO2 B:5
patch DEO2 B:6
patch DEO2 B:10
patch DEO2 B:12
patch DEO1 B:1
patch DEO1 B:3
patch DEO1 B:7
patch DEO1 B:8
patch DEO1 B:9
patch DEO1 B:11
coordpdb B.pdb B
segment SOD {
pdb ions.pdb
}
coordpdb ions.pdb SOD
guesscoord
writepsf psfgen_ions.psf
writepdb psfgen_ions.pdb
END
exit
- Run it with source psfgen_ions.sh, examine log and PDB file.
- Time to hydrate the system : Create a file with the name vmd.script containing the following script (for VMD) :
#
# This is a short script implementing VMD's 'solvate' to prepare
# a fully solvated system for a periodic boundary simulation.
# It also prepares two pdb files needed for implementing restrains
# during the heating-up phase.
#
#
# Make water box
#
solvate psfgen_ions.psf psfgen_ions.pdb -o hydrated -b 1.80 -t 15.0
#
# Here you can add additional ions to emulate higher ionic strength
# conditions. In this case we add 50mM salt, increasing the effective
# ionic strength to approximately 250mM.
#
package require autoionize
autoionize -psf hydrated.psf -pdb hydrated.pdb -is 0.050
#
# Prepare restraints files
#
mol load psf ionized.psf pdb ionized.pdb
set all [atomselect top all]
set sel [atomselect top "nucleic and name P"]
$all set beta 0
$sel set beta 0.5
$all writepdb restrain_P.pdb
set all [atomselect top all]
set to_fix [atomselect top "nucleic and backbone"]
$all set beta 0
$to_fix set beta 1
$all writepdb fix_backbone.pdb
- Run this script via VMD :
- Locate VMD's console (window with the prompt)
- In the console give source vmd.script
- If all goes well you should see your fully hydrated and ionised system on VMD's graphics window.
- To finish type quit in the console.
- Use rasmol or vmd to study your system (ionized.pdb). Make sure that there are no waters or ions accidentally placed inside your molecule's hydrophobic core.
- The last step before running NAMD is to determine the limits (along the orthogonal frame) of the system. A fast method to do this is to use the program pdbset from the CCP4 suite of program. Here it is how :
- Start it : pdbset xyzin ionized.pdb >& pdbset.log
- Edit the file pdbset.log. Somewhere near the end of it you will see something similar to :
Orthogonal Coordinate limits in output file:
Minimum Maximum Centre Range
On X : -37.70 36.29 -0.71 74.00
On Y : -29.96 30.01 0.03 59.97
On Z : -30.39 29.60 -0.39 60.00
- Note these numbers, especially the range (on x,y,z) and position of centre.
- Create a NAMD minimisation and heating-up script with the name heat.namd containing the following :
#
# Input files
#
structure ionized.psf
coordinates ionized.pdb
parameters par_all27_prot_na.inp
paraTypeCharmm on
#
# Output files & writing frequency for DCD
# and restart files
#
outputname output/heat_out
binaryoutput off
restartname output/restart
restartfreq 1000
binaryrestart yes
dcdFile output/heat_out.dcd
dcdFreq 200
#
# Frequencies for logs and the xst file
#
outputEnergies 20
outputTiming 200
xstFreq 200
#
# Timestep & friends
#
timestep 2.0
stepsPerCycle 8
nonBondedFreq 1
fullElectFrequency 2
#
# Simulation space partitioning
#
switching on
switchDist 10
cutoff 12
pairlistdist 13.5
#
# Basic dynamics
#
temperature 0
COMmotion no
dielectric 1.0
exclude scaled1-4
1-4scaling 1.0
rigidbonds all
#
# Particle Mesh Ewald parameters.
#
Pme on
PmeGridsizeX 80 # <===== CHANGE ME
PmeGridsizeY 60 # <===== CHANGE ME
PmeGridsizeZ 60 # <===== CHANGE ME
#
# Periodic boundary things
#
wrapWater on
wrapNearest on
cellBasisVector1 74.00 00.00 00.00 # <===== CHANGE ME
cellBasisVector2 00.00 60.00 00.00 # <===== CHANGE ME
cellBasisVector3 00.00 00.00 60.00 # <===== CHANGE ME
cellOrigin 0.00 0.00 0.00 # <===== CHANGE ME
#
# Fixed atoms for initial heating-up steps
#
fixedAtoms on
fixedAtomsForces on
fixedAtomsFile fix_backbone.pdb
fixedAtomsCol B
#
# Restrained atoms for initial heating-up steps
#
constraints on
consRef restrain_P.pdb
consKFile restrain_P.pdb
consKCol B
#
# Langevin dynamics parameters
#
langevin on
langevinDamping 10
langevinTemp 320 # <===== Check me
langevinHydrogen on
langevinPiston on
langevinPistonTarget 1.01325
langevinPistonPeriod 200
langevinPistonDecay 100
langevinPistonTemp 320 # <===== Check me
useGroupPressure yes
##########################################
# The actual minimisation and heating-up
# protocol follows. The number of steps
# shown below are too small for a real run
##########################################
#
# run one step to get into scripting mode
#
minimize 0
#
# turn off pressure control until later
#
langevinPiston off
#
# minimize nonbackbone atoms
#
minimize 400 ;# <===== CHANGE ME
output output/min_fix
#
# min all atoms
#
fixedAtoms off
minimize 400 ;# <===== CHANGE ME
output output/min_all
#
# heat with Ps restrained
#
set temp 20;
while { $temp < 321 } { ;# <===== Check me
langevinTemp $temp
run 400 ;# <===== CHANGE ME
output output/heat_P
set temp [expr $temp + 20]
}
#
# equilibrate volume with Ps restrained
#
langevinPiston on
run 400 ;# <===== CHANGE ME
output output/equil_P
#
# equilibrate volume without restraints
#
constraintScaling 0
run 2000 ;# <===== CHANGE ME
- You can now give it a quick try with NAMD just to make sure it starts without problems. To avoid overloading machines that are already busy with real calculations you must review the cluster usage as follows :
- Type mosmon from your terminal. You should see something similar to this
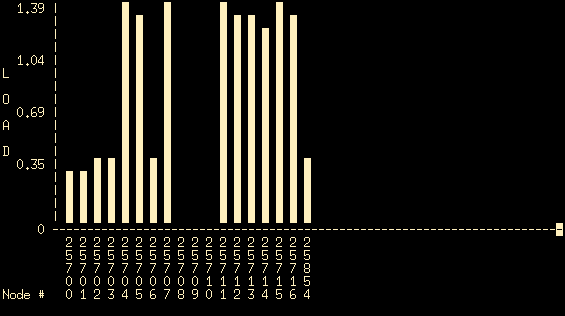
- This graph shows (in real time) the CPU load for each of the nodes that are currently part of the cluster. The horizontal axis shows the various nodes, the vertical axis shows the corresponding load. If all machines are working properly and they are all running the openMosix linux kernel you should see at least 18 nodes (numbered 25700-25716, plus another node with ID 25854). In this case all cluster nodes are running a proper operating system (and can be seen in the mosmon output) and three of these (25708, 25709 and 25710 corresponding to pc08, pc09 and pc10) are idle. So, nodes pc08, pc09 and pc10 are good candidates for performing your tests.
- Type q to stop mosmon and then give qmon (from your terminal). The main Sun Grid Engine GUI window should appear:
- Select "Queue control". This should pop-up the corresponding window :
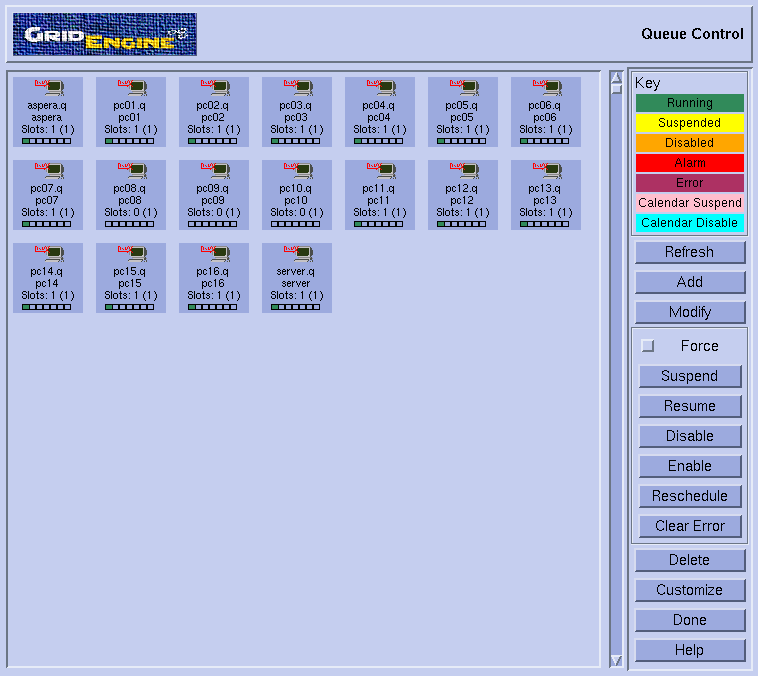
- Note the small green boxes (indicating that a job is running) on all nodes except pc08, pc09 and pc10. So, it does look like we could use these three machines for the tests. Press 'Done' on the queue control window and then exit in the qmon window.
- As a first test lets run NAMD on a single node, e.g. pc09 : login to it with ssh pc9 (use your password to login). Then cd to the directory containing your file : oops, no such file or directory ? That's right : you created the files on your local machine, which probably isn't pc09. So, here comes the need for a cluster-wide filesystem. This is implemented in the form of the /work directory. Proceed as follows :
- Now go back to the directory containing the files you created. Of all the files present in there you only need to copy those few :
cp ionized.p* par_all27_prot_na.inp heat.namd fix_backbone.pdb restrain_P.pdb /work/tmp/my_tests/
- ls and you should see your files (although you are connected to pc9).
- mkdir output will create a subdirectory where the output files from NAMD will be written (during the run).
- Now, go for it : runhome /usr/local/NAMD_2.5/namd2 heat.namd. The runhome command tells the openMosix kernel to run the job on the local machine (not to migrate it away). Once the program performs a couple of minimisation steps stop it with <CTRL-C>. What you should see should be similar to :
Charm++: standalone mode (not using charmrun)
Info: NAMD 2.5 for Linux-i686
Info:
Info: Please visit http://www.ks.uiuc.edu/Research/namd/
Info: and send feedback or bug reports to namd@ks.uiuc.edu
Info:
Info: Please cite Kale et al., J. Comp. Phys. 151:283-312 (1999)
Info: in all publications reporting results obtained with NAMD.
Info:
Info: Based on Charm++/Converse 050612 for net-linux-icc
Info: Built Fri Sep 26 17:33:59 CDT 2003 by jim on lisboa.ks.uiuc.edu
Info: Sending usage information to NAMD developers via UDP. Sent data is:
Info: 1 NAMD 2.5 Linux-i686 1 aspera.cluster.mbg.gr glykos
Info: Running on 1 processors.
Info: 1335 kB of memory in use.
Measuring processor speeds... Done.
Info: Configuration file is heat.namd
TCL: Suspending until startup complete.
Info: SIMULATION PARAMETERS:
Info: TIMESTEP 2
Info: NUMBER OF STEPS 0
Info: STEPS PER CYCLE 8
Info: PERIODIC CELL BASIS 1 74 0 0
Info: PERIODIC CELL BASIS 2 0 60 0
Info: PERIODIC CELL BASIS 3 0 0 60
Info: PERIODIC CELL CENTER 0 0 0
Info: WRAPPING WATERS AROUND PERIODIC BOUNDARIES ON OUTPUT.
Info: WRAPPING TO IMAGE NEAREST TO PERIODIC CELL CENTER.
Info: LOAD BALANCE STRATEGY Other
Info: LDB PERIOD 1600 steps
Info: FIRST LDB TIMESTEP 40
Info: LDB BACKGROUND SCALING 1
Info: HOM BACKGROUND SCALING 1
Info: PME BACKGROUND SCALING 1
Info: MAX SELF PARTITIONS 50
Info: MAX PAIR PARTITIONS 20
Info: SELF PARTITION ATOMS 125
Info: PAIR PARTITION ATOMS 200
Info: PAIR2 PARTITION ATOMS 400
Info: INITIAL TEMPERATURE 0
Info: CENTER OF MASS MOVING? NO
Info: DIELECTRIC 1
Info: EXCLUDE SCALED ONE-FOUR
Info: 1-4 SCALE FACTOR 1
Info: DCD FILENAME output/heat_out.dcd
Info: DCD FREQUENCY 200
Warning: INITIAL COORDINATES WILL NOT BE WRITTEN TO DCD FILE
Info: XST FILENAME output/heat_out.xst
Info: XST FREQUENCY 200
Info: NO VELOCITY DCD OUTPUT
Info: OUTPUT FILENAME output/heat_out
Info: RESTART FILENAME output/restart
Info: RESTART FREQUENCY 1000
Info: BINARY RESTART FILES WILL BE USED
Info: SWITCHING ACTIVE
Info: SWITCHING ON 10
Info: SWITCHING OFF 12
Info: PAIRLIST DISTANCE 13.5
Info: PAIRLIST SHRINK RATE 0.01
Info: PAIRLIST GROW RATE 0.01
Info: PAIRLIST TRIGGER 0.3
Info: PAIRLISTS PER CYCLE 2
Info: PAIRLISTS ENABLED
Info: MARGIN 0.48
Info: HYDROGEN GROUP CUTOFF 2.5
Info: PATCH DIMENSION 16.48
Info: ENERGY OUTPUT STEPS 20
Info: TIMING OUTPUT STEPS 200
Info: FIXED ATOMS ACTIVE
Info: FORCES BETWEEN FIXED ATOMS ARE CALCULATED
Info: HARMONIC CONSTRAINTS ACTIVE
Info: HARMONIC CONS EXP 2
Info: LANGEVIN DYNAMICS ACTIVE
Info: LANGEVIN TEMPERATURE 320
Info: LANGEVIN DAMPING COEFFICIENT IS 10 INVERSE PS
Info: LANGEVIN DYNAMICS APPLIED TO HYDROGENS
Info: LANGEVIN PISTON PRESSURE CONTROL ACTIVE
Info: TARGET PRESSURE IS 1.01325 BAR
Info: OSCILLATION PERIOD IS 200 FS
Info: DECAY TIME IS 100 FS
Info: PISTON TEMPERATURE IS 320 K
Info: PRESSURE CONTROL IS GROUP-BASED
Info: INITIAL STRAIN RATE IS 0 0 0
Info: CELL FLUCTUATION IS ISOTROPIC
Info: PARTICLE MESH EWALD (PME) ACTIVE
Info: PME TOLERANCE 1e-06
Info: PME EWALD COEFFICIENT 0.257952
Info: PME INTERPOLATION ORDER 4
Info: PME GRID DIMENSIONS 80 60 60
Info: Attempting to read FFTW data from FFTW_NAMD_2.5_Linux-i686.txt
Info: Optimizing 6 FFT steps. 1... 2... 3... 4... 5... 6... Done.
Info: Writing FFTW data to FFTW_NAMD_2.5_Linux-i686.txt
Info: FULL ELECTROSTATIC EVALUATION FREQUENCY 4
Info: USING VERLET I (r-RESPA) MTS SCHEME.
Info: C1 SPLITTING OF LONG RANGE ELECTROSTATICS
Info: PLACING ATOMS IN PATCHES BY HYDROGEN GROUPS
Info: RIGID BONDS TO HYDROGEN : ALL
Info: ERROR TOLERANCE : 1e-08
Info: MAX ITERATIONS : 100
Info: RIGID WATER USING SETTLE ALGORITHM
Info: NONBONDED FORCES EVALUATED EVERY 2 STEPS
Info: RANDOM NUMBER SEED 1109584307
Info: USE HYDROGEN BONDS? NO
Info: COORDINATE PDB ionized.pdb
Info: STRUCTURE FILE ionized.psf
Info: PARAMETER file: CHARMM format!
Info: PARAMETERS par_all27_prot_na.inp
Info: SUMMARY OF PARAMETERS:
Info: 250 BONDS
Info: 622 ANGLES
Info: 1049 DIHEDRAL
Info: 73 IMPROPER
Info: 130 VDW
Info: 0 VDW_PAIRS
Info: ****************************
Info: STRUCTURE SUMMARY:
Info: 25117 ATOMS
Info: 17038 BONDS
Info: 9599 ANGLES
Info: 2228 DIHEDRALS
Info: 68 IMPROPERS
Info: 0 EXCLUSIONS
Info: 22 CONSTRAINTS
Info: 176 FIXED ATOMS
Info: 24569 RIGID BONDS
Info: 0 RIGID BONDS BETWEEN FIXED ATOMS
Info: 50254 DEGREES OF FREEDOM
Info: 8647 HYDROGEN GROUPS
Info: 108 HYDROGEN GROUPS WITH ALL ATOMS FIXED
Info: TOTAL MASS = 154535 amu
Info: TOTAL CHARGE = 8.23289e-07 e
Info: *****************************
Info: Entering startup phase 0 with 7301 kB of memory in use.
Info: Entering startup phase 1 with 7301 kB of memory in use.
Info: Entering startup phase 2 with 8984 kB of memory in use.
Info: Entering startup phase 3 with 9184 kB of memory in use.
Info: PATCH GRID IS 4 (PERIODIC) BY 3 (PERIODIC) BY 3 (PERIODIC)
Info: REMOVING COM VELOCITY 0 0 0
Info: LARGEST PATCH (5) HAS 760 ATOMS
Info: Entering startup phase 4 with 12394 kB of memory in use.
Info: PME using 1 and 1 processors for FFT and reciprocal sum.
Creating Strategy 4
Creating Strategy 4
Info: PME GRID LOCATIONS: 0
Info: PME TRANS LOCATIONS: 0
Info: Optimizing 4 FFT steps. 1... 2... 3... 4... Done.
Info: Entering startup phase 5 with 13567 kB of memory in use.
Info: Entering startup phase 6 with 13567 kB of memory in use.
Info: Entering startup phase 7 with 13567 kB of memory in use.
Info: COULOMB TABLE R-SQUARED SPACING: 0.0625
Info: COULOMB TABLE SIZE: 769 POINTS
Info: NONZERO IMPRECISION IN COULOMB TABLE: 2.64698e-22 (712) 2.64698e-22 (712)
Info: NONZERO IMPRECISION IN COULOMB TABLE: 1.26218e-29 (742) 1.57772e-29 (742)
Info: NONZERO IMPRECISION IN COULOMB TABLE: 1.27055e-21 (767) 1.48231e-21 (767)
Info: Entering startup phase 8 with 16909 kB of memory in use.
Info: Finished startup with 21797 kB of memory in use.
TCL: Minimizing for 0 steps
ETITLE: TS BOND ANGLE DIHED IMPRP ELECT VDW BOUNDARY MISC KINETIC TOTAL TEMP TOTAL2 TOTAL3 TEMPAVG PRESSURE GPRESSURE VOLUME PRESSAVG GPRESSAVG
ENERGY: 0 8367.1615 2562.3243 745.3077 0.7495 -86828.1909 2661304.0749 0.0000 0.0000 0.0000 2586151.4269 0.0000 2586151.4269 2586151.4269 0.0000 2770784.8777 2821435.1130 266400.0000 2770784.8777 2821435.1130
TCL: Setting parameter langevinPiston to off
TCL: Minimizing for 400 steps
ETITLE: TS BOND ANGLE DIHED IMPRP ELECT VDW BOUNDARY MISC KINETIC TOTAL TEMP TOTAL2 TOTAL3 TEMPAVG PRESSURE GPRESSURE VOLUME PRESSAVG GPRESSAVG
ENERGY: 0 8367.1615 2562.3243 745.3077 0.7495 -86828.1909 2661304.0749 0.0000 0.0000 0.0000 2586151.4269 0.0000 2586151.4269 2586151.4269 0.0000 2770784.8777 2821435.1130 266400.0000 2770784.8777 2821435.1130
INITIAL STEP: 1e-06
GRADIENT TOLERANCE: 6.07456e+07
ENERGY: 1 8461.3964 2575.7790 745.3495 0.7474 -86744.8887 66834.9357 0.0000 0.0000 0.0000 -8126.6806 0.0000 -8126.6806 -8126.6806 0.0000 62244.7987 65640.3834 266400.0000 62244.7987 65640.3834
ENERGY: 2 8797.5006 2631.6879 745.3945 0.7454 -86710.0767 39177.7886 0.0000 0.0000 0.0000 -35356.9597 0.0000 -35356.9597 -35356.9597 0.0000 33177.5662 35521.3466 266400.0000 33177.5662 35521.3466
ENERGY: 3 10026.8780 2749.8692 745.4941 0.7418 -86681.3159 31642.7422 0.0000 0.0000 0.0000 -41515.5906 0.0000 -41515.5906 -41515.5906 0.0000 24842.4717 28926.6604 266400.0000 24842.4717 28926.6604
ENERGY: 4 16301.5980 3252.9024 745.7321 0.7358 -86621.8424 6474574.1627 0.0000 0.0000 0.0000 6408253.2887 0.0000 6408253.2887 6408253.2887 0.0000 6747235.8916 6748522.4575 266400.0000 6747235.8916 6748522.4575
BRACKET: 6e-06 6.44977e+06 -8.35686e+09 2.51346e+09 1.63163e+09
- Wouldn't it be great to run NAMD a bit faster ? Say, approximately three times faster ? Isn't it a pity to have pc08 and pc10 filtering the dust in the terminal room ? While you are at /work/tmp/my_tests/ create a file with the name NAMD.sh containing the following :
#!/bin/csh -f
#
#
# The name of the job
#
#$ -N My_test
#
# ====> CHANGE ME <====
#
# The parallel environment (mpi_fast) and number of processors (3)
# The options are : mpi_fast : the 9 new machines
# mpi_slow : the 9 older machines
# mpich : all machines on the cluster
#
#$ -pe mpi_fast 3
#
# The version of MPICH to use, transport protocol & a trick to delete cleanly
# running MPICH jobs ...
#
#$ -v MPIR_HOME=/usr/local/mpich-ssh
#$ -v P4_RSHCOMMAND=rsh
#$ -v MPICH_PROCESS_GROUP=no
#$ -v CONV_RSH=rsh
#
# Nodes that can server as master queues
#
#$ -masterq pc01.q,pc02.q,pc03.q,pc04.q,pc05.q,pc06.q,pc07.q,pc08.q,pc09.q,pc10.q,pc11.q,pc12.q,pc13.q,pc14.q,pc16.q
#
# Execute from the current working directory ...
#
#$ -cwd
#
# Standard error and output should go into the current working directory ...
#
#$ -e ./
#$ -o ./
#
# Built nodelist file for charmrun
#
echo "Got $NSLOTS slots."
echo "group main" > $TMPDIR/charmlist
awk '{print "host " $1}' $TMPDIR/machines >> $TMPDIR/charmlist
cat $TMPDIR/charmlist
#
# ====> CHANGE ME <====
#
# The name of the NAMD script file is defined here (heat.namd) as well as
# the name of the job's log file (LOG)
#
/usr/local/NAMD_2.5/charmrun /bin/runhome /usr/local/NAMD_2.5/namd2 ++nodelist $TMPDIR/charmlist +p $NSLOTS heat.namd > LOG
- This script is a parallel NAMD job submission script for the Sun Grid Engine. All you have to do is : qsub NAMD.sh (from the /work/tmp/my_tests/ directory). You should see a message containing your job's ID.
- Type qstat : you should see your job and its status changing from qw to t to r.
- Use mosmon to confirm that the previously idle machines are indeed doing something useful.
- Use qmon to review cluster usage, and to see your job in Job Control/Running Jobs.
- tail -f LOG should show you the growing log file from your job.
- Type mknamdplot LOG and then click Draw to see a collection of graphs showing the evolution of quantities like the system's total energy, pressure, temperature, bond and angle energies, … :
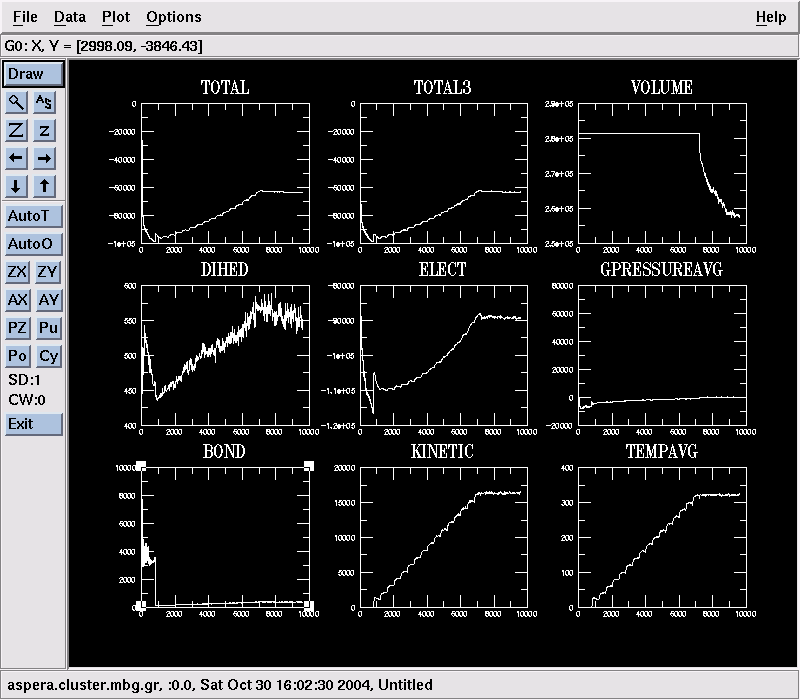
- The heating-up NAMD job will take 2-3 hours to finish. Let it do so. But if you'd rather not : qdel JobID will kill your job (or you can use the qmon GUI).
- When the job is done review the files that have been created in /work/tmp/my_tests/output/. You should see something like
-rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:19 equil_P.coor
-rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:19 equil_P.vel
-rw-rw-r-- 1 glykos glykos 222 Oct 30 15:19 equil_P.xsc
-rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:16 heat_P.coor
-rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:13 heat_P.coor.BAK
-rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:16 heat_P.vel
-rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:13 heat_P.vel.BAK
-rw-rw-r-- 1 glykos glykos 153 Oct 30 15:16 heat_P.xsc
-rw-rw-r-- 1 glykos glykos 153 Oct 30 15:13 heat_P.xsc.BAK
-rw-rw-r-- 1 glykos glykos 2029503 Oct 30 15:36 heat_out.coor
-rw-r--r-- 1 glykos glykos 14613396 Oct 30 15:36 heat_out.dcd
-rw-rw-r-- 1 glykos glykos 2029502 Oct 30 15:36 heat_out.vel
-rw-rw-r-- 1 glykos glykos 227 Oct 30 15:36 heat_out.xsc
-rw-rw-r-- 1 glykos glykos 3364 Oct 30 15:36 heat_out.xst
-rw-rw-r-- 1 glykos glykos 2029502 Oct 30 14:25 min_all.coor
-rw-rw-r-- 1 glykos glykos 2029501 Oct 30 14:25 min_all.vel
-rw-rw-r-- 1 glykos glykos 152 Oct 30 14:25 min_all.xsc
-rw-rw-r-- 1 glykos glykos 2029502 Oct 30 14:19 min_fix.coor
-rw-rw-r-- 1 glykos glykos 2029501 Oct 30 14:19 min_fix.vel
-rw-rw-r-- 1 glykos glykos 152 Oct 30 14:19 min_fix.xsc
-rw-rw-r-- 1 glykos glykos 608836 Oct 30 15:31 restart.coor
-rw-rw-r-- 1 glykos glykos 608836 Oct 30 15:22 restart.coor.old
-rw-rw-r-- 1 glykos glykos 608836 Oct 30 15:31 restart.vel
-rw-rw-r-- 1 glykos glykos 608836 Oct 30 15:22 restart.vel.old
-rw-rw-r-- 1 glykos glykos 229 Oct 30 15:31 restart.xsc
-rw-rw-r-- 1 glykos glykos 228 Oct 30 15:22 restart.xsc.old
- Play time : from the output/ directory give vmd ../ionized.psf heat_out.dcd
- You can now start preparing the equilibration-production run :
- cp heat_out.coor heat_out.vel heat_out.xsc ../../equi/
- cp ionized.p* par_all27_prot_na.inp ../equi/
- Create a NAMD equilibration script with the name equi.namd containing the following :
#
# Input files
#
structure ionized.psf
coordinates heat_out.coor
velocities heat_out.vel
extendedSystem heat_out.xsc
parameters par_all27_prot_na.inp
paraTypeCharmm on
#
# Output files & writing frequency for DCD
# and restart files
#
outputname output/equi_out
binaryoutput off
restartname output/restart
restartfreq 1000
binaryrestart yes
dcdFile output/equi_out.dcd
dcdFreq 200
#
# Frequencies for logs and the xst file
#
outputEnergies 20
outputTiming 200
xstFreq 200
#
# Timestep & friends
#
timestep 2.0
stepsPerCycle 8
nonBondedFreq 1
fullElectFrequency 2
#
# Simulation space partitioning
#
switching on
switchDist 10
cutoff 12
pairlistdist 13.5
#
# Basic dynamics
#
COMmotion no
dielectric 1.0
exclude scaled1-4
1-4scaling 1.0
rigidbonds all
#
# Particle Mesh Ewald parameters.
#
Pme on
PmeGridsizeX 80 # <===== CHANGE ME
PmeGridsizeY 60 # <===== CHANGE ME
PmeGridsizeZ 60 # <===== CHANGE ME
#
# Periodic boundary things
#
wrapWater on
wrapNearest on
wrapAll on
#
# Langevin dynamics parameters
#
langevin on
langevinDamping 1
langevinTemp 320 # <===== Check me
langevinHydrogen on
langevinPiston on
langevinPistonTarget 1.01325
langevinPistonPeriod 200
langevinPistonDecay 100
langevinPistonTemp 320 # <===== Check me
useGroupPressure yes
firsttimestep 9600 # <===== CHANGE ME
run 4000 ;# <===== CHANGE ME
- Prepare a submission script for SGE, say NAMD.sh :
#!/bin/csh -f
#
#
# The name of the job
#
#$ -N test_equi
#
# The parallel environment (mpi_fast) and number of processors (9) ...
#
#$ -pe mpi_fast 3
#
# The version of MPICH to use, transport protocol & a trick to delete cleanly
# running MPICH jobs ...
#
#$ -v MPIR_HOME=/usr/local/mpich-ssh
#$ -v P4_RSHCOMMAND=rsh
#$ -v MPICH_PROCESS_GROUP=no
#$ -v CONV_RSH=rsh
#
# Nodes that can server as master queues
#
#$ -masterq pc01.q,pc02.q,pc03.q,pc04.q,pc05.q,pc06.q,pc07.q,pc08.q,pc09.q,pc10.q,pc11.q,pc12.q,pc13.q,pc14.q,pc16.q
#
# Execute from the current working directory ...
#
#$ -cwd
#
# Standard error and output should go into the current working directory ...
#
#$ -e ./
#$ -o ./
#
# Built nodelist file for charmrun
#
echo "Got $NSLOTS slots."
echo "group main" > $TMPDIR/charmlist
awk '{print "host " $1}' $TMPDIR/machines >> $TMPDIR/charmlist
cat $TMPDIR/charmlist
#
# Ready ...
#
/usr/local/NAMD_2.5/charmrun /bin/runhome /usr/local/NAMD_2.5/namd2 ++nodelist $TMPDIR/charmlist +p $NSLOTS equi.namd > LOG
![[Home]](/wiki.png)